Can Transformers Learn the Language of the Microbiome?

Drugs that you take interact with a complex microbial ecosystem in your gut. Trillions of microorganisms call the human digestive tract home and interact with one another and with their human host. This microbial world can directly modify drug structure (Ref. 1–4), influence host immune responses (5,6), and alter pharmacokinetics (4,7). Currently, most drug development programs choose to ignore the gut microbiome because a reliable method to predict how it impacts drugs does not exist.
That’s the problem we’re working on at Outpost Bio: making human microbiology computable. A big part of that challenge is building AI models that can learn meaningful representations of microbial communities from data. Here we present a series of GPT-2-style language models pretrained on one of the largest microbiome datasets ever assembled, introduce a new benchmark to measure what those models learn, and run scaling analyses to understand the effects of larger data and models. The results show that pretraining on large, unlabeled datasets improves performance on a range of microbiome prediction tasks, enables microbiome foundation models to outperform traditional baselines at obtainable dataset sizes, and unlocks the full potential of larger models. This work establishes a strong foundation, but it’s only the beginning of what scalable microbiome modeling can unlock, and we’re excited to see how the community builds on it.
Treating microbiomes as sequences
To make this work, we needed to distill complex microbiome data into a structure that machines can read. A microbiome sample is represented as a list of microbial taxa and their relative abundances: which organisms are present, and how much of each. Following the approach in Zhang et al. (Ref 8), we order the taxa by their normalized abundance scores and feed the resulting sequence into a transformer. The model’s job during pretraining is the same as any generative language model: predict the next token.
The pretraining corpus is one of the largest aggregations of publicly available microbiome data to date. We scraped over 500,000 microbiome data points from the MGnify (Ref 9) database, spanning amplicon, shotgun metagenomic, and assembly data from samples across environments ranging from marine and soil to the human gut and skin.
To increase data retention, we introduced a genus with a fallback tokenization strategy. In many microbiome studies, especially those using 16S amplicon sequencing, you can’t always classify a microbial read down to species or even genus level. Previous approaches (8, 10, 11) either discarded those reads or forced a single taxonomic level. We tokenize taxa by using the genus label, falling back to the next specific taxon when a genus isn’t assigned. In practice, this means you keep data that would otherwise be thrown away. This approach increased our dataset size by approximately 200,000 datapoints.
A standardized benchmark to allow model comparison
One of the things that’s been holding back progress in microbiome ML is the lack of a standardized benchmark. Different papers evaluate model quality on different tasks (8, 10, 11), making it hard to compare approaches or measure real progress.
This work introduces a standardized benchmark consisting of a curated set of eight prediction tasks drawn from four independent datasets, designed to closely reflect real-world microbiome applications (Table 1). The tasks span the types of problems a microbiome model should be able to solve.
• Biome classification: predicting whether a sample came from marine, soil, gut, or other environments (Tasks 1 and 2). Taken from held-out MGnify (9) samples that are not used in pretraining.
• Drug–microbiome interactions: identifying which individual a perturbed community came from, whether there was a drug treatment, and which drug class (Tasks 3, 4, and 5). Taken from an investigation on the effect of drugs on stool-derived communities (12).
• Drug degradation: predicting how fast a community breaks down a compound, the only regression task in the benchmark (Task 6), using data from a high-throughput screen of drug degradation by gut microbiomes (13).
• Infant development: predicting age and delivery mode from an infant’s gut microbiome (Tasks 7 and 8), using data from a longitudinal study of infant gut microbiomes (14).
Classification tasks are scored using macro-averaged F1, the unweighted mean of the F1 score for each class, which penalizes models that predict the majority class. The F1 score is a single number that measures how well you balance not missing real positives (recall) and not making false positives (precision), with values closer to 1 indicating a better model. The regression task is evaluated using R2 (coefficient of determination), which measures how much of the variation in the target variable the model explains; values closer to 1 indicate a better fit.
Task | Dataset | Type | Metric |
| Richardson et al. (9) | Classification | F1-macro |
| Richardson et al. (9) | Classification | F1-macro |
| Classification | F1-macro | |
| Classification | F1-macro | |
| Regression | R2 | |
| Mastrorilli et al. (13) | Classification | F1-macro |
| Roswall et al. (14) | Classification | F1-macro |
| Roswall et al. (14) | Classification | F1-macro |
Table 1. Benchmark task summary. Classification tasks are scored using macro-averaged F1; the regression task (Task 6) is scored using R2.
Does scale matter?
We trained nine models ranging from 6 million to 170 million parameters and evaluated them on our benchmark (Table 2). All share the same tokenizer, context length (512 tokens), and training procedure, so differences in performance can be attributed solely to model capacity.
Model | Layers | Hidden dim | Heads |
6M-MGM | 8 | 256 | 8 |
6M | 8 | 256 | 4 |
10M | 8 | 320 | 5 |
18M | 10 | 384 | 6 |
29M | 12 | 448 | 7 |
45M | 14 | 512 | 8 |
79M | 16 | 640 | 10 |
85M-gpt2-small | 12 | 768 | 12 |
170M | 24 | 768 | 12 |
Table 2. GPT-2 model configurations used in the scaling study.
We benchmarked our pretrained GPT-2 models against linear (logistic regression for classification, ridge regression for regression tasks) and random forest (RF) classical approaches, both trained on simple bag-of-taxa abundance profiles using the same vocabulary as the transformer models.
We also compared our models to MGM (Ref. 8), an existing microbiome foundation model trained on 260,000 MGnify samples using the same GPT-2 architecture. To provide an apples-to-apples comparison between our larger pretraining corpus and prior work, we included the 6M-MGM model architecture, which exactly matched the MGM model. Although we aimed to include other recent microbiome foundation models (10,11), the lack of publicly available weights made this impossible. Finally, to assess the effect of pretraining itself, we tested versions of each transformer architecture with randomly initialized weights.
Three findings stand out.
1. Pretraining on public data improves performance on task-specific data
Every pretrained model outperformed every non-pretrained transformer and both classical baselines (random forest and logistic/ridge regression) on the overall benchmark score (Figure 1). The improvement is consistent across all models, pointing to a systematic advantage rather than a chance outcome.
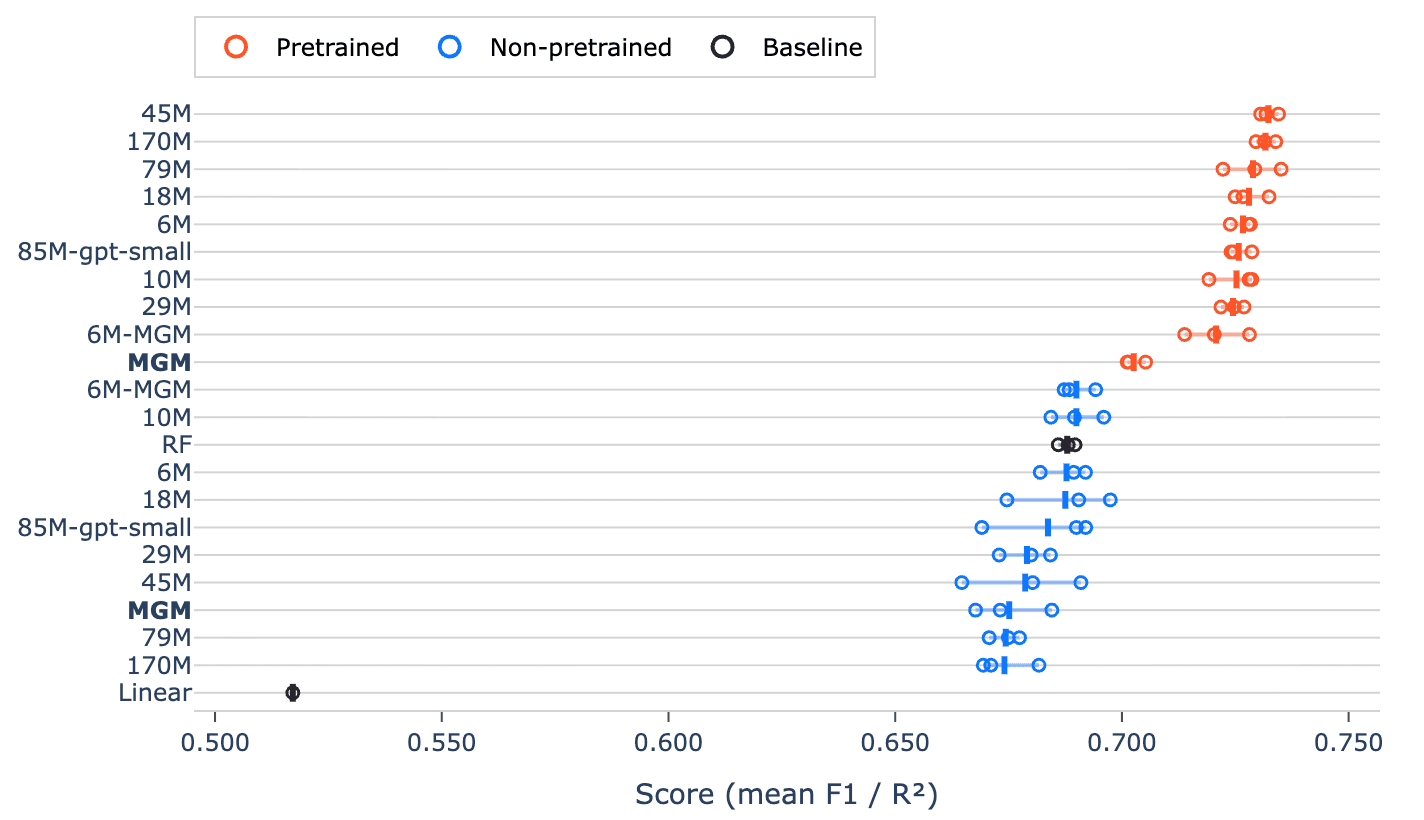
Figure 1. Overall benchmark scores for all models. Pretrained models consistently outperform non-pretrained models and the baselines.
What’s notable here is that even relatively small pretrained models surpass larger or equally sized models trained from scratch, and that the effect of pretraining is bigger than scaling model size. In other words, pretraining on large-scale, unlabeled microbiome data appears to matter more than simply scaling parameters. The fact that pretrained transformers also outperform well-established baselines such as random forests (a traditionally strong performer on tabular biological data) underscores how much useful structure is captured during pretraining.
A particularly informative comparison is between 6M-MGM and the original MGM model. This demonstrates that, when the architecture is held constant, the model trained on more data and with improved tokenization performs better. We can further assess the improved tokenization scheme by comparing the non-pretrained MGM and 6M-MGM models. Here, the only difference is the improved tokenization scheme, and we see that our 6M-MGM model still outperforms MGM. This illustrates that how we represent these communities is a key modeling decision.
When we break down the results for each benchmarking task, we find that pretraining consistently improves performance, though the degree of improvement varies by task (Figure 2). This reinforces a key point: self-supervised pretraining is an effective way to learn general-purpose microbiome representations that transfer well across a wide range of downstream tasks. Additionally, we see that for benchmarking task 4 (drug treatment) and task 7 (infant age), simpler baseline models still outperform all transformer-based approaches. Thus, for small datasets, the added complexity of transformers does not necessarily improve performance.
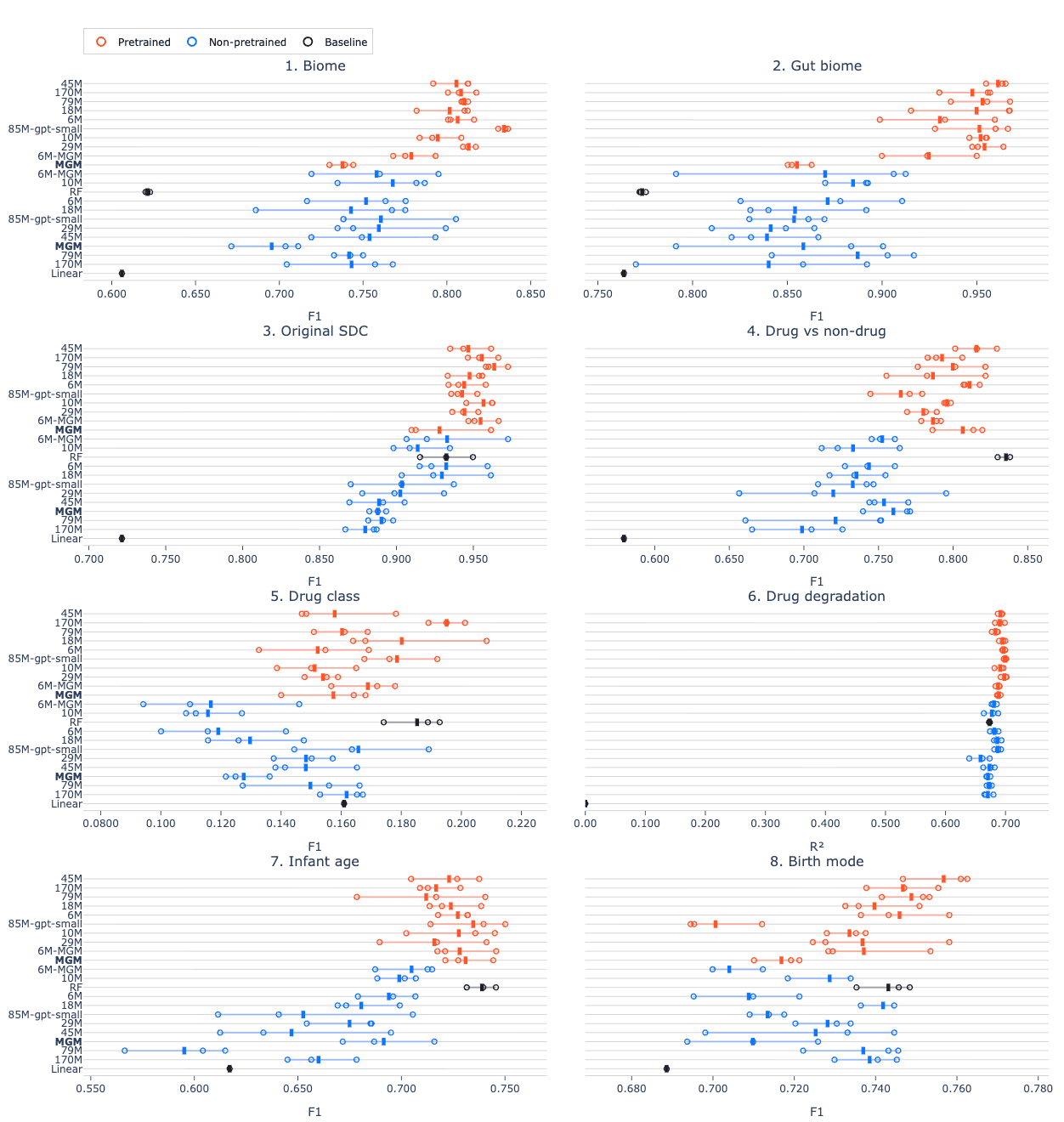
Figure 2. Score per task for all models. Pretrained models consistently outperform non-pretrained models, but for some tasks, the RF baseline outperforms transformer models.
Taken together, these results show that self-supervised pretraining fundamentally improves the quality of learned representations, enabling models to generalize better across the diverse set of tasks in the benchmark. This has exciting implications for the future of microbiome modeling.
2. Transformers beat baselines - with enough data
Using the tasks in our benchmark, we also examined how performance differs with training set size, focusing on where pretrained transformers begin to outperform classical baselines (Figure 3). A clear pattern emerges: the crossover point is around 10,000 labeled examples. Below this threshold, methods like random forest remain highly competitive and can even be preferable given their simplicity and strong performance in low-data regimes. But once you move beyond ~10k samples, pretrained transformers consistently pull ahead, and the gap widens as more data becomes available.
This behavior highlights an important trade-off. Classical models are more efficient and robust on small, limited datasets, but they cannot fully leverage larger datasets. On the other hand, pretrained transformers are better able to learn the additional signal as the dataset size grows, because they start from a much stronger initialization and benefit from both scale and prior representation learning.
Crucially, 10,000 samples is no longer an unrealistic target. In vitro microbiome screening is unlocking large datasets. Notably, the dataset exploring how diverse microbiomes metabolize drugs, characterized by Mastrorilli et al. (Ref. 13, used for task 6 in our benchmark), contained 15,419 samples, of which 9,282 were used for training. Many research groups are operating at or beyond this scale, not only in vitro but also with large longitudinal population-level cohorts.
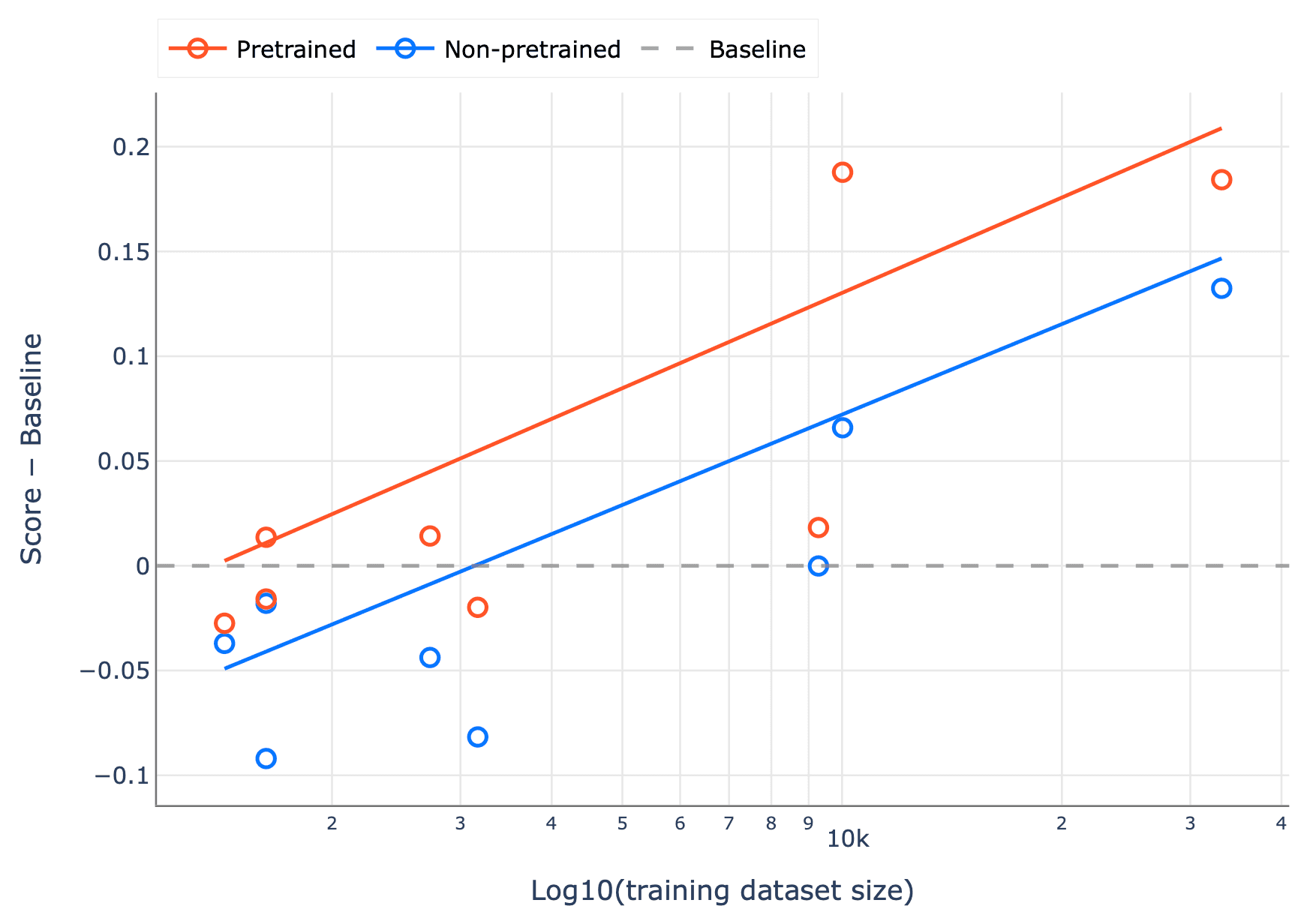
Figure 3. The difference in mean score across the three replicates relative to the RF baseline, plotted against the size of the training dataset for each task. At 10k and above, all pretrained transformers are better than the baseline. Solid lines are OLS lines of best fit.
3. Pretraining unlocks scaling
When we plot benchmark performance against model size, two very different scaling behaviors emerge (Figure 4). For pretrained models, the trend is clear and intuitive: larger models perform better. As capacity increases, these models can capture more structure in the data and translate it into improved downstream performance.
In contrast, non-pretrained models show the opposite pattern. Increasing model size without pretraining hurts performance, with larger randomly initialized transformers tending to do worse. This likely reflects the difficulty of training high-capacity models from scratch on relatively limited labeled datasets: they have more parameters to tune, but not enough signal to guide them effectively, leading to poorer generalization.
The practical implication is straightforward: scaling model size only pays off if it’s paired with pretraining. Pretraining allows the model to leverage its additional capacity by starting from a meaningful representation of the data.
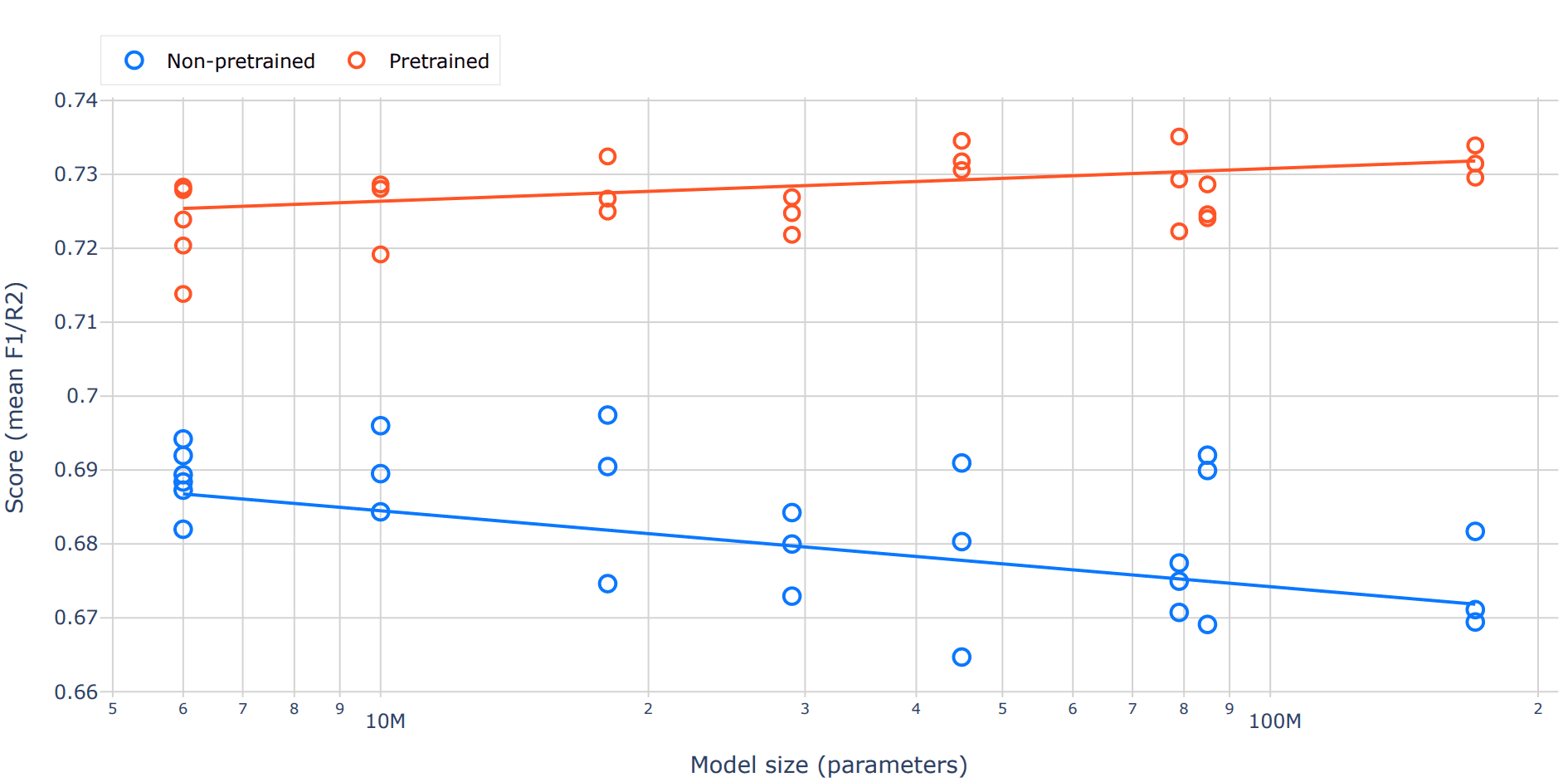
Figure 4. Benchmark performance as a function of model size for pretrained and non-pretrained transformer models. Each point represents the score from an individual replicate across tasks, with lines indicating linear trends. Pretrained models show consistent improvements with increasing parameter count, while non-pretrained models exhibit a decline in performance at larger scales, highlighting the importance of pretraining for effective model scaling.
Why this matters
First, we have shown that self-supervised pretraining on large microbiome datasets produces representations that transfer across a wide range of downstream tasks. In biology, where generating high-quality labeled data in the wet lab is expensive, the ability to learn from large public datasets is especially valuable.
Second, our scaling analysis provides important context when deciding where to invest compute. In natural language processing, the relationship between model size, data, and performance is well understood. For the microbiome, this is the first systematic evidence that similar principles apply to pretrained models.
Third, we will open-source our pretraining corpus to enable others to train and explore their own models.
Fourth, we will release our standardized and reproducible benchmark that gives the microbiome ML community a much-needed way to compare methods on common ground.
Finally, we will open-source our best-performing pretrained models alongside the data and benchmark, so the community can build directly on these results, whether that’s fine-tuning on new tasks, probing the representations, or using them as a starting point for novel investigations. We believe the field moves faster when data, benchmarks, and models are shared.
What's still missing
This is just the starting point. Our benchmark currently focuses on gut and environmental tasks. We will soon expand to include other samples, such as skin, oral, and respiratory microbiomes. We're also planning to expand the baseline comparisons beyond random forests and linear regression to include gradient boosting and alternative neural architectures, giving a fuller picture of where foundation models truly pull ahead. Notably, two other microbiome foundation models couldn't be included because their weights aren't publicly available (Ref. 10,11) which reinforces why open science matters for this field to move forward.
On the modeling side, there's plenty of room to push further. Our current 512-token context window handles the vast majority of samples, but scaling context length for high-diversity communities should be investigated. We're also exploring whether richer representations of relative abundance can outperform rank-ordering. The literature is split on this (10,11), and we think it's a question worth settling with better data. Empirically, we observed minimal differences in sequence length distributions and total token counts between genus-level and species-level (with fallback) tokenization strategies, and therefore proceeded at the genus-level. Nonetheless, this choice may limit taxonomic resolution, and the impact of more precise tokenization schemes remains an open question. We also plan to explore a broader range of open-source architectures beyond GPT-2, as newer models may offer meaningful gains in this domain. And all models here use a causal, left-to-right objective; bidirectional pretraining could meaningfully improve performance on non-generative tasks.
Beyond public datasets, we are the only company in this space generating proprietary functional data in our own wet lab. We are developing stool-derived in vitro microbiome models that represent the natural variation of human microbiomes. These microbial ecosystems enable controlled intervention studies to address how drugs/nutrients perturb the microbiome and in turn, how the microbiome impacts these interventions. Notably, these systems are scalable and support rapid iteration, allowing cost-effective data generation that will empower the next generation of our models.
The bottom line
We've assembled a large, diverse pretraining corpus with a practical tokenization improvement, introduced a reproducible benchmark the community can build on, and run a scaling analysis showing that pretraining is both effective and necessary for microbiome foundation models. Our work demonstrates that the field has crossed an inflection point: there is now sufficient data to train large-scale foundation models that outperform traditional approaches. The remaining bottleneck is the generation of high-quality, task-specific training data. At Outpost Bio, we are addressing this by tightly coupling machine learning with wet lab experiments in a lab-in-the-loop framework, enabling rapid iteration between model development and data generation. We now know that machine learning can be applied to microbiome data. Now, we're asking, “How do we build models reliable enough to inform drug development decisions?” This work lays the foundation, and we are continuing to build on it. We are releasing these resources in the spirit of open science because we believe that broad collaboration is the fastest path to meaningful progress in this field.
Outpost Bio is building frontier AI models for human microbiology. We combine high-throughput functional assays on human-derived microbial communities with causal machine learning to predict how drugs and nutrients interact with the microbiome. If you’re working on oral drug development, metabolism prediction, or microbiome-aware R&D, we’d love to talk.
Trepka, K. R. et al. Expansion of a bacterial operon during cancer treatment ameliorates drug toxicity. Preprint at https://doi.org/10.1101/2024.06.04.597471 (2024).
Hou, Y. et al. <em>Bacteroides intestinalis</em> mediates the sensitivity to irinotecan toxicity via tryptophan catabolites. Gut gutjnl-2024-334699 (2025) doi:10.1136/gutjnl-2024-334699.
Mehta, R. et al. GUT MICROBIAL METABOLISM OF 5-ASA IS PROSPECTIVELY ASSOCIATED WITH TREATMENT FAILURE IN PATIENTS WITH IBD. Inflamm. Bowel Dis. 29, S57–S58 (2023).
Saqr, A., Cheng, S., Al‐Kofahi, M., Staley, C. & Jacobson, P. A. Microbiome‐Informed Dosing: Exploring Gut Microbial Communities Impact on Mycophenolate Enterohepatic Circulation and Therapeutic Target Achievement. Clin. Pharmacol. Ther. 118, 1477–1488 (2025).
Petrelli, F. et al. Clinical Evidence for Microbiome-Based Strategies in Cancer Immunotherapy: A State-of-the-Art Review. Medicina (Mex.) 61, 1595 (2025).
Melissa Anahi Chan Verdugo et al. The Interplay Between the Gut Microbiome and Immunotherapy in Gastrointestinal Malignancies: Mechanisms, Clinical Implications, And Therapeutic Potential. Int. J. Med. Sci. Clin. Res. Stud. 5, 890–896 (2025).
Drevland, O. M. et al. Microbiome-derived reactivation of mycophenolate explains variations in enterohepatic recirculation in kidney transplant recipients. Microbiome 13, 169 (2025).
Zhang, H. et al. MGM as a Large-Scale Pretrained Foundation Model for Microbiome Analyses in Diverse Contexts. Adv. Sci. n/a, e13333 (2026).
Richardson, L. et al. MGnify: the microbiome sequence data analysis resource in 2023. Nucleic Acids Res. 51, D753–D759 (2023).
Pope, Q., Varma, R., Tataru, C., David, M. & Fern, X. Learning a deep language model for microbiomes: the power of large scale unlabeled microbiome data. Preprint at https://doi.org/10.1101/2023.07.17.549267 (2023).
Medearis, N. A., Zhu, S. & Zomorrodi, A. R. BiomeGPT: A foundation model for the human gut microbiome. bioRxiv https://doi.org/10.64898/2026.01.05.697599 (2026) doi:10.64898/2026.01.05.697599.
Shi, H. et al. Nutrient competition predicts gut microbiome restructuring under drug perturbations. Cell 0, (2025).
Mastrorilli, E., Herd, P., Rey, F. E., Goodman, A. L. & Zimmermann, M. Linking interpersonal differences in gut microbiota composition and drug biotransformation activity. bioRxiv 2026.01.21.700809 (2026) doi:10.64898/2026.01.21.700809.
Roswall, J. et al. Developmental trajectory of the healthy human gut microbiota during the first 5 years of life. Cell Host Microbe 29, 765-776.e3 (2021).
Work on a problem that matters.
Our mission is ambitious, and our standards are high. Our culture is built on intellectual honesty, deep ownership, reliably doing what we say we will do. If you are a builder who thrives on candid feedback and are ready to do the best work of your career, we would love to meet you.
An open invitation to researchers.
We’re building AI to predict how interventions impact the human microbiome and will be contributing foundational datasets and models back to the scientific community. We’re seeking collaborators with rich pre/post-intervention datasets of the microbiome. We'll offer partners rich insights to existing datasets.
© Outpost Bio 2025
Design by The Sourdough
