Squeezing more out of 16S: a gut-specific reference for functional imputation

Microbiome teams keep coming back to the same unglamorous question: what assay can we afford to run, and what are we giving up when we choose it?
For DNA-based profiling, the usual split is 16S versus shotgun metagenomics. With 16S, you sequence a marker gene. The fixed regions make it easy to target, and the variable regions give you a decent read on taxonomy: broadly, who is in the sample. Shotgun metagenomics is a different proposition. You sequence all accessible DNA, which gives better taxonomic resolution and, more importantly for us, a way to ask what genes and functions are present.
The trade-off is familiar. 16S is cheaper, cleaner to analyse, and much easier to scale. Shotgun tells you more. In an early-stage tech-bio setting, the ratio of cost to information is a useful metric for assay selection.
One way people have tried to get more out of 16S is functional imputation. The basic move is to map the 16S sequence you observed onto a reference phylogeny, infer the likely genome behind it, and then borrow the functions annotated on that genome or its relatives. PICRUSt2 [1] is the standard tool here. It places 16S features into a reference tree and uses hidden-state prediction to infer gene families or pathways.
PICRUSt2 works well in broad terms. The possible weakness, for our purposes, is its reference. Much of the human gut microbiome i.e one of the key ecosystems Outpost is focused on, is full of organisms that have never been grown in a lab, let alone genomically characterized. If the real relatives of a gut sequence are missing from the reference, that sequence gets placed near more distant, usually culturable taxa. The imputed gene repertoire can then miss enzymes carried by the actual lineage, including context-specific adaptations that may be the most interesting part of the sample.
A natural question follows: can a gut-specific reference make 16S functional imputation more useful?
The Unified Human Gastrointestinal Genome catalogue (UHGG) [2], distributed through MGnify [3,4], is an obvious candidate. Version 2.0 of the UHGG catalogue contains 289,232 prokaryotic genomes from cultured isolates and gut metagenomes, clustered into 4,744 species representatives. Most of those species, 80.8% in v2.0, have no cultured representative. That is exactly the missing space where a broad isolate-heavy reference might struggle.
We built a simple version of the idea and then tried to make the comparison hard to fool.
The pipeline and the test
The pipeline has very few moving parts. We used samples with 16S readouts from the Human Microbiome Project (HMP1) [7]. Each 16S feature (in this case an HMP1 sequence defining an operational taxonomic unit or OTU), is aligned to UHGG 16S sequences with VSEARCH [5]. We assign it to the closest UHGG species representative. From that species representative, we read out the enzyme repertoire using eggNOG-mapper [6] EC annotations. Then we sum enzyme calls across the sample, weighted by 16S abundance. We call the resulting method UHGG-EC.
There is one design choice worth flagging early. If an OTU ties across several UHGG species representatives, we assign the union of their enzymes. That makes the method recall-first by construction. It also means we should expect lower precision. This is not a small implementation detail; it shows up in the results.
For a useful test, we needed the same stool samples profiled by both 16S and shotgun WGS. HMP1 [7] gives us that. We used 113 stool samples with paired 16S V3-V5 and shotgun data. The WGS side came from HMP’s HUMAnN2 [8] reprocessing, which maps reads to a protein reference and regroups them to EC numbers. That gives a per-sample list of enzymes detected from whole-genome data, with a median of roughly 1,270 enzymes per sample.
Then we asked a plain question: using only the 16S data from those same samples, how many of the WGS-detected enzymes does each method recover? We compared UHGG-EC against a standard PICRUSt2-to-EC pipeline.
This is a better test than a toy benchmark. The samples are real human guts. The truth assay is the one we would rather not pay for every time. The readout is the actual functional label we want downstream.
There is also a nuisance that matters more than you would hope. The three EC profiles are produced through different annotation routes. HUMAnN uses a UniRef50-to-EC table. UHGG-EC uses eggNOG EC annotations. PICRUSt2 uses KEGG KO-to-EC mappings. Those vocabularies overlap, but they are not identical. If one method can emit an EC number and the other cannot, a naive recall comparison will credit biology when some of the difference is really bookkeeping.
We therefore report two versions of the result: the naive full-universe comparison, and the cleaner comparison restricted to EC numbers both pipelines are capable of producing.
Result 1: the gain survives vocabulary control, but it gets smaller
Over the full union of EC numbers that any method can emit, UHGG-EC recovers 82% of the WGS-truth enzymes per sample. PICRUSt2 recovers 73%. That is an 8.6 percentage-point recall advantage for UHGG-EC, with paired bootstrap p < 0.001.
Taken at face value, that looks like the headline.
Once we restrict the comparison to the EC vocabulary shared by both 16S pipelines, the advantage drops to 4.3 points: 93.2% recall for UHGG-EC versus 88.9% for PICRUSt2, again with p < 0.001. In other words, about half of the apparent win comes from the annotation layer. eggNOG gives UHGG-EC broader EC coverage than the KEGG KO-to-EC mapping used here for PICRUSt2.
The annotation advantage is still useful in practice; a user running the pipeline really does get those extra EC calls. It should just be kept separate from the biological claim. For gut-specific placement, the shared-vocabulary result is the number to lean on.
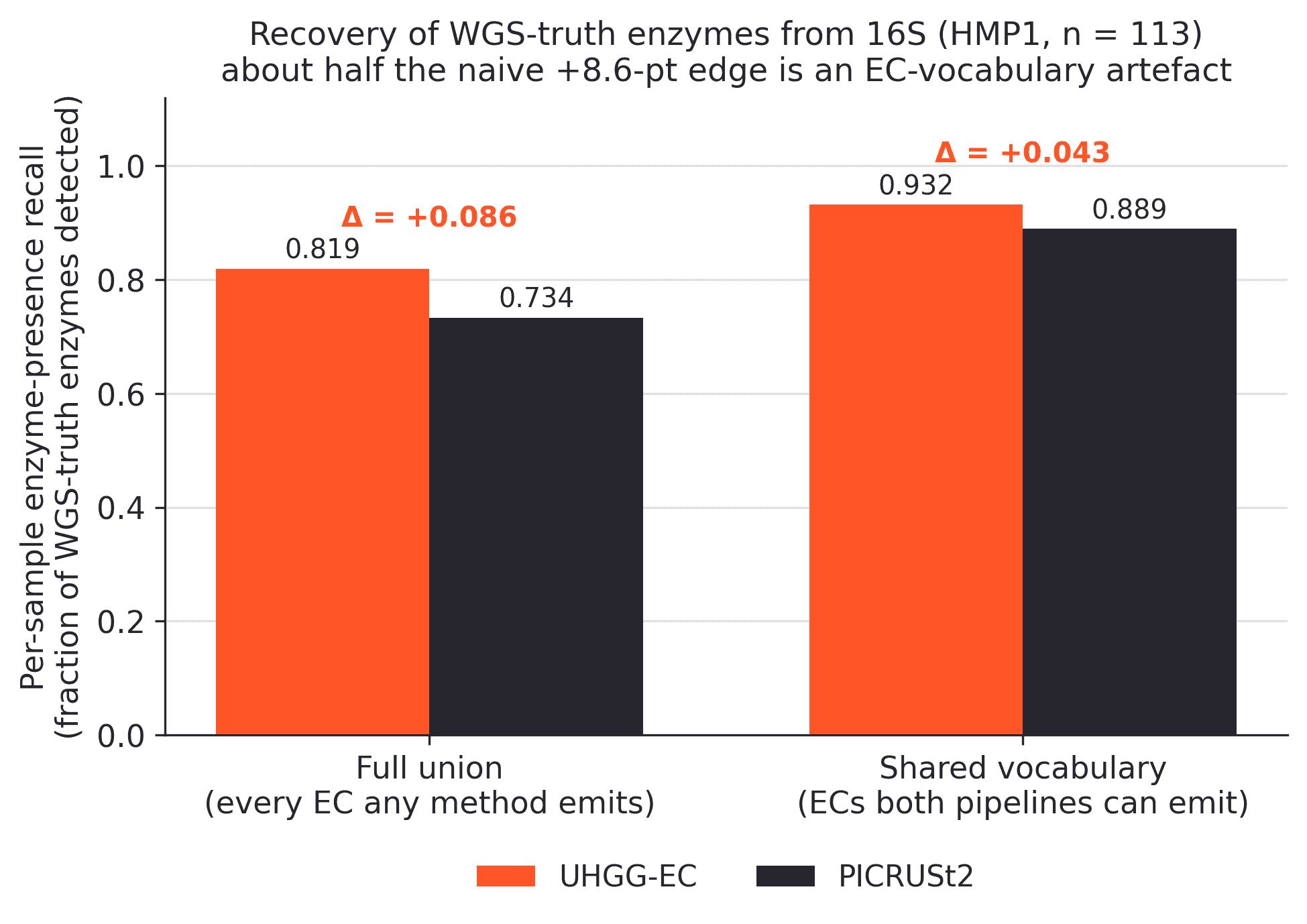
Recovery of WGS-truth enzymes from 16S (113 paired HMP1 stool samples). Measured over the full union of EC numbers any method can emit (left), UHGG-EC leads PICRUSt2 by 8.6 points of presence-recall.
Restricting to the EC vocabulary both pipelines can express (right) cuts the gap to 4.3 points, showing how much of the naive advantage comes from annotation coverage rather than placement.
The more interesting part is where the remaining gain sits. When truth enzymes are binned by their prevalence across samples, the recall gap gets wider as the enzymes get rarer.
|
|
|
|
|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(shared-vocabulary universe, 113 samples)
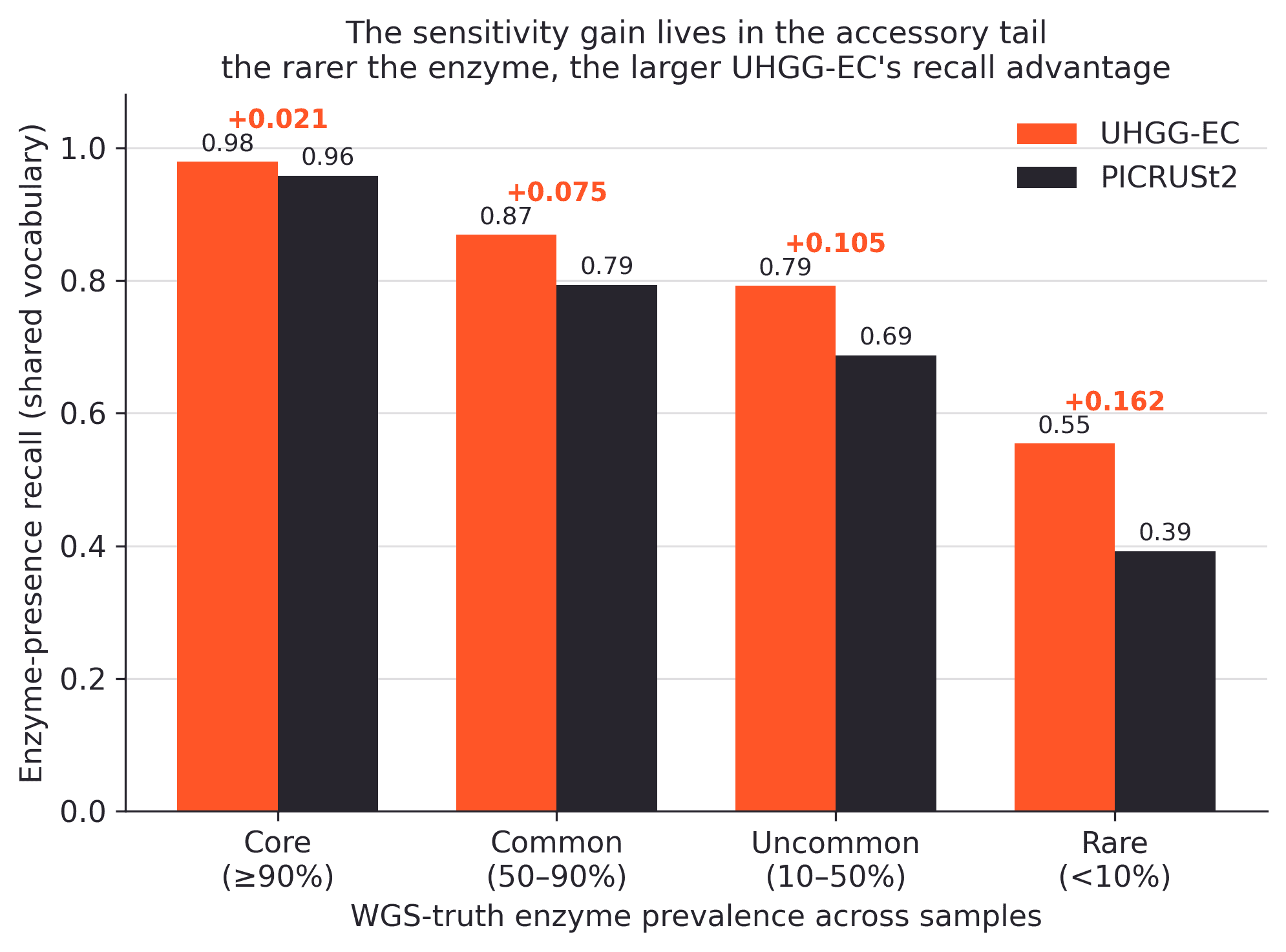
The sensitivity gain lives in the accessory tail. Enzyme-presence recall by how prevalent each truth enzyme is across samples, in the shared-vocabulary universe. Both methods are close to ceiling on the conserved core. UHGG-EC’s advantage grows as enzymes get rarer, reaching a 16-point gain on the rare, sample-specific tail.
At the conserved core, there is not much room to improve. Both methods already find almost everything. UHGG-EC earns its keep in the accessory tail: the rarer, more sample-specific enzymes where 16S imputation is most likely to fail and where gut-specific sequence context should matter most.
For us, the tail matters more than the core. Conserved housekeeping functions are not where most discovery hypotheses come from. The more useful signal is in the enzymes that vary across strains, hosts, diets, drugs, and ecological niches.
Result 2: UHGG-EC behaves like a recall-first screen
The same results that make UHGG-EC look good on recall also show its limitations. It calls more enzymes per sample than PICRUSt2: about 1,510 versus 1,310, against a truth set of about 1,020 on the shared vocabulary. A wider net catches more real enzymes. It also catches more false positives.
Precision drops accordingly. UHGG-EC reaches 0.63 precision on the shared vocabulary, compared with 0.69 for PICRUSt2. The extra calls are not pure junk: about 23% of UHGG-EC-only calls are confirmed by WGS, in the same general range as PICRUSt2-only calls, which are confirmed about 26% of the time. Still, the cost is real. You get more of the repertoire, and you pay for it with specificity.
A matched-budget test tells a similar story. If each method is allowed to name exactly as many enzymes as the WGS truth contains, their top-K sets are close. PICRUSt2 is ahead by about 2.0 points on the shared vocabulary. UHGG-EC is ahead by about 2.6 points on the full union. That sign flip is a useful warning: neither method has a decisive ranking advantage per call. UHGG-EC’s recall gain mostly comes from calling more enzymes, with a non-trivial fraction of the additional calls landing in real accessory functions.
Abundance is a separate issue, and PICRUSt2 does better there. On enzymes called by both methods, PICRUSt2’s relative abundance estimates track WGS abundance more closely in magnitude, with log-scale Pearson correlation of 0.63 versus 0.51 for UHGG-EC. No surprise there: PICRUSt2 has a hidden-state model, copy-number estimates, and confidence weighting. UHGG-EC, in this version, is closer to a binary gene-presence lookup summed by 16S abundance.
UHGG-EC does have a higher whole-profile rank correlation, with Spearman 0.65 versus 0.60, but that is mostly a presence/absence effect across many shared zeros. It should not be read as better quantitative calibration.
The practical interpretation is fairly narrow. UHGG-EC is useful when missing a candidate enzyme is more costly than carrying extra candidates forward. It is a discovery screen. For calibrated pathway abundance, PICRUSt2 remains the safer choice, or at least the method to keep in the comparison.
Result 3: the average gut is annoyingly strong
There is a baseline that makes every 16S functional method look less impressive. If you ignore the sample’s 16S profile and simply predict the average gut enzyme profile, you recover a lot of the truth.
In a matched-budget setting, where each method is allowed to name the same number of enzymes as the truth, this average-gut prior recovers about 89% of enzymes on the full universe. UHGG-EC recovers 71%, and PICRUSt2 recovers 69%. On the shared vocabulary the pattern is similar; the prior reaches 91%.
A large conserved functional core will do that. If most samples share the same central machinery, a generic prior gets a long way without reading the sample at all.
The prior breaks exactly where the biology gets more interesting. On the rare, sample-specific tail, it recovers under 2%. The 16S-based methods recover roughly 18% of that same tail, around nine times more.
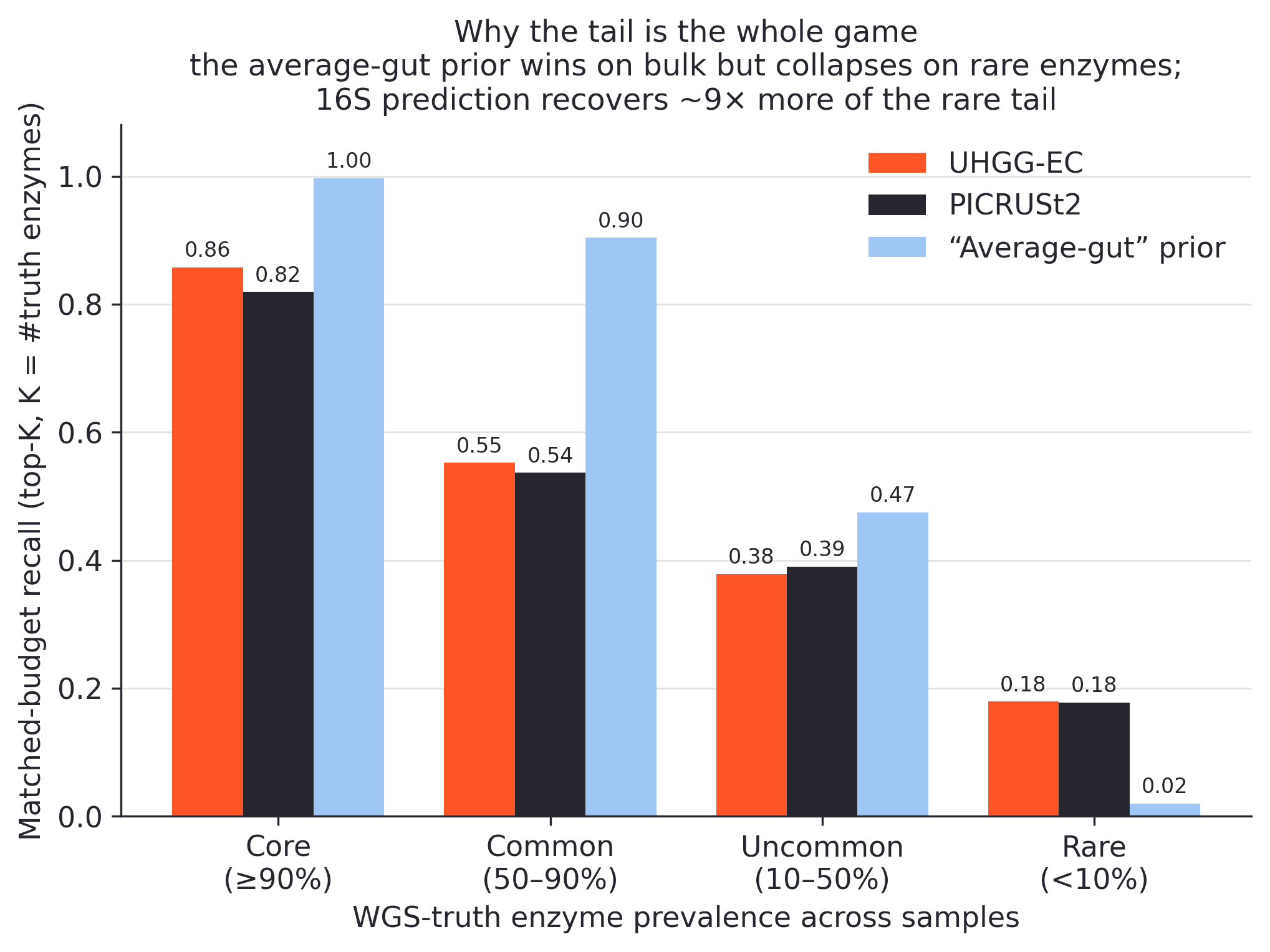
Why the tail is the whole game. Matched-budget recall, where each method is allowed to name exactly K enzymes per sample, broken down by enzyme prevalence. An average-gut prior dominates the conserved core but recovers under 2% of the rare, sample-specific tail. The 16S-based methods recover about 18% of that tail.
This changes the way to think about the method. 16S imputation is not very valuable for rediscovering enzymes that almost every gut already has. A prevalence prior handles those. The value of reading the sample is in the variable tail, even though recall there is still modest in absolute terms.
The same region is where UHGG-EC has its largest edge over PICRUSt2.
Result 4: the uncultured genomes help less than the simple story suggests
The tempting explanation for UHGG-EC’s gain is the uncultured majority of UHGG. If 80.8% of UHGG species have no cultured representative, perhaps the method wins because it can represent gut lineages that PICRUSt2’s isolate-heavy reference misses.
That mechanism is present, but it is not the main driver in this cohort.
In the HMP1 stool samples, uncultured taxa account for a median of only 4% of the 16S abundance that maps to UHGG. Most of the mappable signal is still close to cultured species. In addition, about 94% of UHGG-EC’s correct enzyme recoveries are also reachable through a cultured relative. The bulk of the recall gain is therefore not coming from enzymes that exist only in uncultured dark matter.
A more prosaic explanation fits the data better. UHGG-EC reads the full gene complement from gut genomes directly, while PICRUSt2 imputes a more conservative set from a broader phylogenetic neighbourhood. That helps for cultured and uncultured taxa alike.
The uncultured signal does show up where it should. Among enzymes recovered by UHGG-EC and missed by PICRUSt2, the rate of being recoverable only through uncultured lineages is about seven times higher than among enzymes both methods recover, with p < 0.001. The dark-matter story is real; it is just smaller than the pitch would imply for HMP1.
A reasonable expectation is that the effect would be larger in under-represented populations, where a greater fraction of the gut microbiome is absent from isolate collections. This dataset cannot establish that. It gives a prediction to test next.
What this means for assay choice
If the budget only allows 16S, UHGG-EC gives a measurable gain in functional recovery for human gut samples. After controlling for EC vocabulary, the gain is 4.3 points of recall, and it is concentrated in the accessory enzyme tail.
UHGG-EC does not turn 16S into shotgun metagenomics. Abundance calibration remains weak, and precision falls. Those are not footnotes; they determine where the method should and should not be used.
For a practitioner who wants calibrated pathway abundances, PICRUSt2 is still the better starting point. Running both methods may be sensible because they fail differently and are cheap once the reads exist. For a practitioner who wants a broad list of candidate enzymes to filter downstream, UHGG-EC is more attractive. The method is doing what a discovery front end should do: preserve signal that would otherwise be missed.
For us, the discovery use case is the reason to care. A high-recall enzyme list from cheap 16S can point to proteins worth folding, docking, expressing, or assaying. Each call traces back to a gene sequence in a gut genome, often one not represented by a cultured isolate. The list is noisy, but downstream filters are supposed to deal with noise. They cannot rescue a candidate that never made it into the list.
Closing the loop with assays
The weaknesses of UHGG-EC are also a map of where new data would help. Its EC calls come from homology-based annotation, often on draft genomes or MAGs. The method also over-calls by design, because we chose a recall-first operating point. Both problems are most acute in the same region: accessory enzymes from poorly characterised gut lineages.
Wet-lab data has leverage there.
Culturing gut organisms turns anonymous or low-confidence lineages into cleaner references. Enzyme assays replace a predicted EC label with a measured activity on a specific protein. Binding or functional screens can separate plausible candidates from dead ends. In this setup, the assay is not an afterthought. It is the precision filter that lets a broad 16S-derived candidate list become useful.
The nice part is that the assay output is compatible with the reference itself. UHGG is built from genomes and annotations. A genome with experimentally measured functions is the same kind of object, just with better labels. Those labels can feed back into a proprietary sequence-to-function map focused on the enzymes where public annotation is weakest.
The workflow is therefore iterative rather than one-off. Cheap 16S produces candidate functions. Structure and sequence models help prioritise them. The lab tests the shortlist. The results improve the reference for the next round.
The strategy is targeted depth. It will not re-annotate hundreds of thousands of genomes wholesale, and it adds little on the conserved core where all methods already perform well. The conserved core is not the bottleneck. The bottleneck is the accessory, drug-relevant slice of functional space where sample-specific information matters and public labels are thin.
In practice, the reference narrows the search space, and the assay work decides which predictions are worth carrying forward.
Caveats
HUMAnN2 is a reference-based truth. It maps WGS reads to a pre-clustered protein catalogue, ChocoPhlAn plus UniRef50, and therefore has its own reference biases. A stricter comparison would assemble, bin, and annotate MAGs per sample. This is a useful independent comparator, not ground truth in the philosophical sense.
The EC-vocabulary mismatch is a real confound. That is why the shared-vocabulary result is the main biological comparison. eggNOG’s broader EC coverage is useful in practice, but it should not be mistaken for better gut-specific placement.
HMP1 is Western and relatively culture-dominant. The uncultured-taxa advantage is small here. It may be larger in cohorts whose gut diversity is less well represented by isolate collections, but that needs to be tested directly.
PICRUSt2 version matters. We used the meta-analysis-bundled PICRUSt2 v2 KO output for comparison with prior work. A fresh v2.5+ run could move the gap, although the shared-vocabulary recall result is unlikely to vanish entirely.
Union tie-handling pushes UHGG-EC toward high recall. Assigning the union of tied species’ pangenomes to an OTU inflates the enzyme list. That behaviour is useful for a generate-then-filter workflow and inappropriate for a precision-first one.
References
Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, Huttenhower C, Langille MGI. PICRUSt2 for prediction of metagenome functions. Nature Biotechnology. 2020;38(6):685-688. doi:10.1038/s41587-020-0548-6
Almeida A, Nayfach S, Boland M, Strozzi F, Beracochea M, Shi ZJ, Pollard KS, Sakharova E, Parks DH, Hugenholtz P, Segata N, Kyrpides NC, Finn RD. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nature Biotechnology. 2021;39(1):105-114. doi:10.1038/s41587-020-0603-3
Gurbich TA, Almeida A, Beracochea M, Burdett T, Burgin J, Cochrane G, Raj S, Richardson L, Rogers AB, Sakharova E, Salazar GA, Finn RD. MGnify Genomes: A Resource for Biome-specific Microbial Genome Catalogues. Journal of Molecular Biology. 2023;435(14):168016. doi:10.1016/j.jmb.2023.168016
Richardson L, Allen B, Baldi G, Beracochea M, Bileschi ML, Burdett T, Burgin J, Caballero-Perez J, Cochrane G, Colwell LJ, Curtis T, Escobar-Zepeda A, Gurbich TA, Kale V, Korobeynikov A, Raj S, Rogers AB, Sakharova E, Sanchez S, Wilkinson DJ, Finn RD. MGnify: the microbiome sequence data analysis resource in 2023. Nucleic Acids Research. 2023;51(D1):D753-D759. doi:10.1093/nar/gkac1080
Rognes T, Flouri T, Nichols B, Quince C, Mahe F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. doi:10.7717/peerj.2584
Cantalapiedra CP, Hernandez-Plaza A, Letunic I, Bork P, Huerta-Cepas J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Molecular Biology and Evolution. 2021;38(12):5825-5829. doi:10.1093/molbev/msab293
Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207-214. doi:10.1038/nature11234
Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, Huttenhower C. Species-level functional profiling of metagenomes and metatranscriptomes. Nature Methods. 2018;15(11):962-968. doi:10.1038/s41592-018-0176-y
Work on a problem that matters.
Our mission is ambitious, and our standards are high. Our culture is built on intellectual honesty, deep ownership, reliably doing what we say we will do. If you are a builder who thrives on candid feedback and are ready to do the best work of your career, we would love to meet you.
An open invitation to researchers.
We’re building AI to predict how interventions impact the human microbiome and will be contributing foundational datasets and models back to the scientific community. We’re seeking collaborators with rich pre/post-intervention datasets of the microbiome. We'll offer partners rich insights to existing datasets.
© Outpost Bio 2025
Design by The Sourdough
